Understanding chemical reactions involving nuclear quantum effects such as tunneling needs the time evolution of a molecular wave function (nuclear wave packet). However, in conventional methods, the number of grid points or basis functions used to express the wave packet increases exponentially with the system size. To avoid this problem, we developed a practical approximate method for efficient wave packet expansion in terms of a Gaussian basis set. The localized nature of Gaussian functions enables a flexible selection of their arrangement.
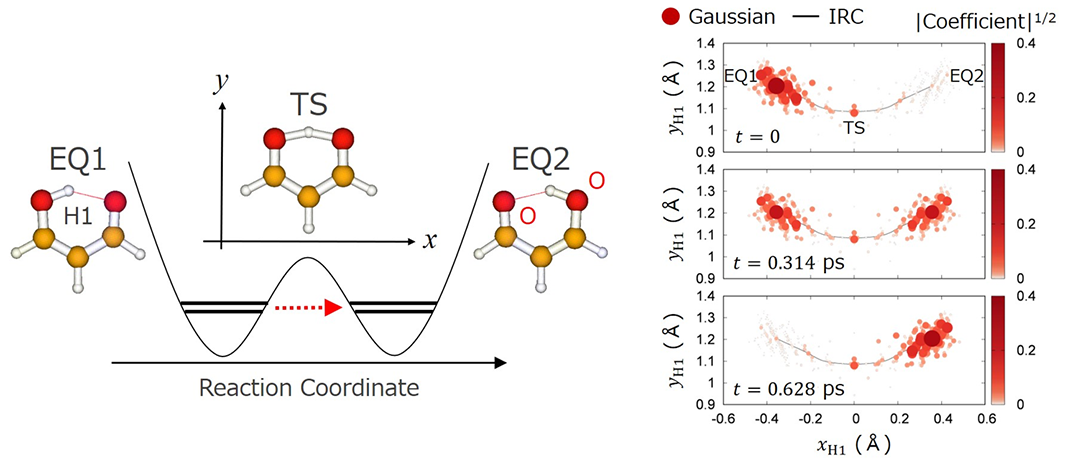
In our method, Gaussians are mainly placed around reaction pathways such as the intrinsic reaction coordinate (IRC) connecting equilibrium structures (EQs) through transition states (TSs), which reduces the expansion length. For hydrogen tunneling in malonaldehyde (see the left panel of the figure below), we calculated the time evolution of a superposition of the lowest two vibrational eigenstates initially localized around EQ1. The right panel shows SBG expansion coefficients at the central positions of H1. Transfer of H1 to EQ2 is visualized in the snapshot at t = 0.63 ps.